This page describes the standard evidence development pathway followed by examples of how evidence development pathways may change when incorporating real-world evidence (RWE) into the pathway. Examples include the use of early pragmatic trials, the introduction of adaptive pathways, or the use of real-world data (RWD) in trial planning.
Standard evidence development pathway
Medicines in development progress through a sequence of trials of increasing size and complexity. Trials are designed to minimise risk to participants and to optimise unbiased reporting of safety and efficacy. Trial designs are agreed in advance with clinical experts, statisticians and regulatory authorities. Marketing authorisation (regulatory approval) is primarily driven by the results of the confirmatory (phase 3) trial(s). The planned sequence of trials for a medicine for a particular indication is usually called the clinical development plan. This plan may be modified as new trials results become available. The biggest, and often riskiest, investment decision for a manufacturer is usually the decision to proceed with the phase 3 trial. Until recently the differing requirements of regulatory and reimbursement/health technology assessment (HTA) agencies has had limited impact on the clinical development plan and the design of individual trials. Figure 1 below describes the standard pathway. Following this, alternative evidence development pathways using RWE are described.
Figure 1. Standard pathway
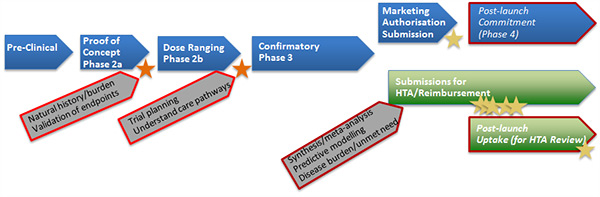

- Effectiveness estimates (for HTA) come from phase 2b and phase 3 trials, supplemented as appropriate with meta-analyses and modelling of long-term outcomes.
- Scientific advice processes may be used to assess the clinical development plan (before phase 2b) or specific study designs (before phase 3).
- Reimbursement assessments are undertaken at a country or healthcare ‘payer’ level, starting immediately after marketing authorisation.
- Conventional uses of real-world data (RWD) in the standard pathway are to describe the natural history and burden of disease, to understand and quantify treatment pathways, to validate new endpoints for use in trials and to develop risk equations for use in predictive models.
- RWD (in the form of observational studies) may also be collected as post-approval commitments (regulatory) or for HTA reviews (reimbursement).
There are current initiatives to improve alignment between regulatory and reimbursement processes, and to improve coordination across HTAs. For more information on these, see Key Important Initiatives.
Modified evidence development pathway: early pragmatic trial
A ‘phase 3b’ pragmatic trial can be introduced to the evidence development pathway when there are concerns that the confirmatory (explanatory) phase 3 trial will not provide sufficient data to on relative effectiveness to reimbursement decision makers. In this case, the pragmatic trial starts while the phase 3a trial is running, and continues in parallel to the marketing authorisation process. Some interim study results may be available in time for HTA/reimbursement assessments, although the pragmatic trial may continue beyond this. The trial will need to be approved by regulatory authorities, who may be particularly interested in collection of data on safety. Alternatively, a pragmatic trial may be conducted (slightly later) in response to a request from an HTA/reimbursement agency for more data on the real-world effects of a medicine, after the medicine has received marketing authorisation (regulatory approval).
Figure 2. Early pragmatic trial pathway
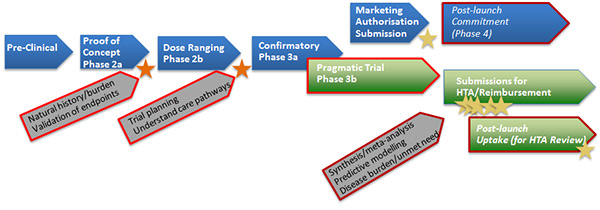
- It may be challenging to set up and report the results of a pragmatic trial in time for reimbursement submissions immediately after marketing authorisation – there is a risk of delaying these submissions.
- If relative effectiveness estimates are to be based on the (phase 3b) pragmatic trial, this may have implications for the design of the phase 3a confirmatory trial. For example, it may be designed to have a shorter duration based on a purely ‘efficacy’ endpoint.
- Avoiding inconsistencies between the trial results (phase 2b, 3a, 3b) is desirable.
- Statistical adaptations of study results (such as analyses and modelling) may still be needed to render the pragmatic trial results relevant to other care settings.
- Real-world data will still be needed for ‘standard evidence development pathway’.
Modified evidence development pathway: adaptive pathway
Adaptive pathways, also known as medicines adaptive pathways to patients (MAPPs) or adaptive licensing, are being tested in medicines for which there is a high unmet need, where clinical trials expose patients who are unlikely to benefit to possible harms, or where traditional trial data collection is challenging (for example, for rare diseases). In adaptive pathways, marketing authorisation is granted iteratively within the existing regulatory framework in successive stages, initially to a restricted population but then incrementally expanding to broader populations as more data become available to confirm the balance of benefit vs harm. Initial authorisation is based on trials with efficacy endpoints considered as surrogates for effectiveness. Successive reimbursement assessments may then follow the expanding marketing authorisation. Adaptive pathways place much greater emphasis on the collection of RWD following early approval, as well as early involvement of stakeholders in helping to shape the pathway.
Figure 3. Adaptive pathway
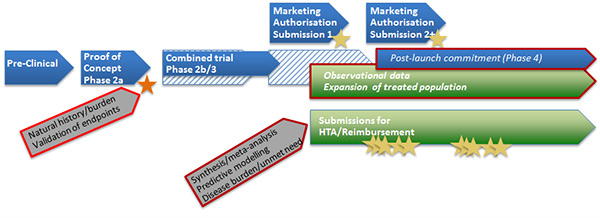

- In this pathway earlier, but constrained, marketing authorisation (regulatory approval) is obtained, based potentially on a shorter clinical trial programme.
- Early regulatory approval may be followed by early, but also constrained, HTA submissions.
- Further rounds of submissions to regulatory and reimbursement/HTA authorities follow as new data become available. These subsequent assessments and reviews therefore take on a greater significance in the pathway.
- Observational RWD collected after the first regulatory approval may be used in subsequent submissions to regulatory authorities for expanded marketing authorisation.
- There is an enhanced opportunity to collect RWD to inform successive submissions and reviews by HTA agencies.
- It may be possible to unify certain activities, especially observational studies, to meet the needs of the different stakeholders.
There are a number of initiatives considering the use of adaptive pathways, such as the EMA Adaptive pathways to patients, IMI ADAPT-SMART, and the New Drug Development Paradigms (NEWDIGS) programme. The NEWDIGs programme has contributed to an initiative to test the impact of adaptive licensing, including the use of real-world evidence (for more information, see Simulation of Alternative Evidence Development Pathways).
For other key related initiatives see Key Important Initiatives.
Modified evidence development pathway: use of RWD in trial planning
Early analyses (and meta-analyses) of effectiveness data for comparator therapies from trials and from observational studies may help refine the design of conventional (explanatory) phase 3 trials when a standard evidence development pathway is followed. This may help to refine/target the trial population, or to reduce the sample size and/or study duration required.
Figure 4. RWD in trial planning pathway
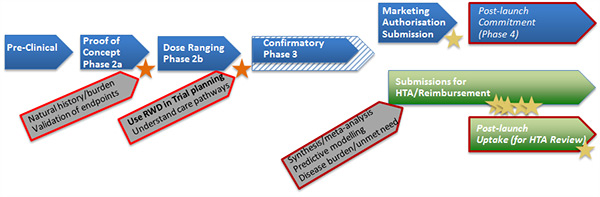
- It is possible that using RWD in designing the phase 3 trial may reduce the time to marketing authorisation.
- Implications for reimbursement/HTA submissions need to be assessed carefully: possibly modify the inclusion or exclusion criteria.
- RWD will still be needed for other purposes, for example for post-approval commitments as described in the ‘standard evidence development pathway’.